
05.01.17
Jeffrey Domsic, Michael Loewenstein and Jeff Van Komen • The Procter & Gamble Company;
Beth Ann Browne • The Dow Chemical Company; Tony Rook • The Sherwin-Williams Company;
Cecilia McGough • Lanxess Corporation
For the Consumer Specialty Products Association Microbiology Preservative Subcommittee
This is the second article of a two-part series dedicated to helping consumer, household and industrial (CH&I) product manufacturers produce products that are uncompromised by microbial contamination (see part 1). CH&I products are often classified as “micro susceptible,” meaning that they are prone to microbial contamination due to a variety of factors that can include compositional features such as high water content and naturally-derived raw materials (RM). In part I of the series, five key components were described for consideration during the CH&I product manufacturing process, including facility and equipment design, cleaning, sanitization, monitoring and documentation.
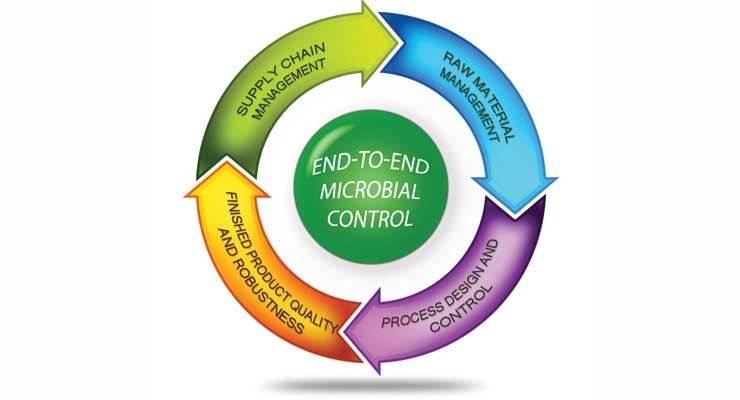
In this article, these components are further described within the context of the manufacturing process in key areas:
These areas represent the end-to-end model in the manufacturing process. General recommendations along with some specific guidance will be provided for consideration by the reader to improve microbial control in manufacturing systems. CH&I products and the processes to manufacture them are varied and distinct; therefore, the suggestions and tips presented herein may not be directly applicable to all systems and products. It is recommended that the appropriate subject matter experts be consulted in areas such as process engineering, regulatory and microbiology before implementing any manufacturing changes at your facility.
Supply Chain and Raw Material Management
Monitoring: Maintaining microbial integrity of each of the raw materials (RM) is key for effective microbial control of the finished product. A microbiological risk assessment of all RM should be performed to determine material susceptibility to their potential and actual level of microbial contamination. Material sampling, testing and specifications can be established to appropriately manage the risk of a RM lifecycle. Proper RM management programs should be considered as important as finished product testing. When combined, RM, environmental and finished product testing data can establish confidence for product release. Additional information on RM testing can be found in Microbiological Quality of Raw Material General Guideline (MGG 003.1 2014).1
RM control strategies begin with adequate supply chain management of incoming RM. While RM suppliers do not have direct oversight for CH&I product facilities and operations, their attention to RM microbial control will impact the CH&I producing company. A variety of RM suppliers service the CH&I product industry, but they differ notably from one another in their understanding of microbial risks to their materials and contamination potential. For this reason, final product manufacturers must gather sufficient information to effectively assess suppliers. It is common for the CH&I company to conduct a safety and quality inspection of the RM facility prior to final purchase. Supplier monitoring may be accomplished via periodic reviews of documentation and/or physical audits of facilities. During an audit, any issues outlined here can be evaluated and corrective actions can be put into place prior to receipt of the RM. Assessing supply chain logistics is often crucial as it can significantly impact the microbial contamination risk of RM. Examples of these factors include environmental conditions like weather, material transit time (including potential long term storage in transit), transfer equipment and the use of non-dedicated vessels. Certain RM are often considered critical for production or present greater microbial contamination risk; therefore, key suppliers and risk mitigation practices are typically established to protect the supply chain.
Documentation: Typically, materials which are susceptible to microbial contamination are tested by suppliers, and results are added to its certificates of analysis (CoA). A CoA may offer a level of assurance to use the RM without additional microbiological testing, but further risks may be accrued during shipment. Understanding the frequency of microbial testing reported on a CoA by suppliers is also important. A best practice of microbiological quality assurance for the supply chain is to evaluate materials upon receipt and prior to use by finished product producers with proper documentation of test data for traceability, tracking and trending the data.
For any RM testing, proper procedures which include the training of those taking samples should be set. For example, procedures for handling, opening, closing/sealing containers through the sampling process should be in place. Also imperative in the documentation process is the ability to react when a RM is determined to be out of specification. Standard Operating Procedures should be in place to guide these processes.
Cleaning: RM can be delivered in a variety of packaging types. RM that are bulk delivered in reusable containers present different challenges than those supplied in disposable packaging. Even drums are frequently resused. Single-use containers should be used whenever feasible and practical. Proper procedures for maintaining reused packing are critical for microbial control from the RM supplier location to the manufacturing facility. Reused containers dedicated to a specific RM are preferred; however, RM can be frequently transported in bulk quantities in reusable containers shared with other materials. Proper cleaning of containers, according to a validated procedure, is needed between uses to maintain the quality of the material, and documentation of the process should be available upon receipt of material(s). Within manufacturing systems, cleaning of RM storage handling equipment is vital. Many RM, such as silicones, can build up within the manufacturing system. RM systems that are not properly cleaned create environments conducive to microbial growth, raising risks for microbial contamination.
Sanitization: RM may be transported and stored in a variety of conditions, and decisions must be made about the frequency of sanitization of the storage vessels, and the maximum amount of time allowed between sanitization and vessel filling. Large reusable transport vessels should be sanitized prior to being filled with a microbiologically susceptible RM. However, not all vessel types can be sanitized, such as lined rail cars, which require other risk mitigation methods to be identified and implemented, such as confined entry inspection to verify cleaning.
Documentation of the sanitization process or alternative risk mitigation steps should be included in the delivery documentation. Upon receipt at the manufacturing site, some materials may be stored for extended periods in the shipped vessels while other RM may be transferred into large storage tanks. Sanitization frequencies can vary based on how long the RM is exposed to the system. For example, a RM with rapid turnover in storage systems would pose different risks than the same material stored for long periods with infrequent use.
Facility and Equipment Design: Upon receipt, RM must be offloaded at the manufacturing site. Specific procedures should be in place for this process, noting that some materials will need special considerations. It is preferred to use in-house offloading hoses instead of transporter hoses for liquids as the recipient has greater control over proper cleaning, sanitization and storage of the in-house hoses between uses. Likewise, the location for unloading bulk samples needs adequate protection for microbial control.
RM quality within the manufacturing site after offloading encompasses a large portion of the manufacturing process, and the fundamentals of process control and design outlined in the next section will cover precautions for equipment used in conjunction with RM. It should be noted that exterior storage of RM, often done because of large usage volumes, is an area that requires special considerations, but that area is beyond the scope of this article.
Process Design & Control
Monitoring: It is essential to monitor manufacturing systems to ensure they are in control. Typical programs may include monitoring of environmental air, bulk storage tanks, in-process mixes, etc. Regulatory bodies give little to no prescriptive guidance on breadth or frequency of sampling and monitoring. Rather, it is left to the company to develop a sampling plan that is sufficient to demonstrate consistent control of each system. However, USP <1116> Microbiological Control and Monitoring of Aseptic Processing Environments,2 and USP <1231> Water for Pharmaceutical Purposes3 are good resources to learn about principles that should be considered when developing monitoring programs. The same types of considerations should be taken into account for all monitoring programs including those for bulk storage tanks and in-process equipment. Some factors may include microbiological susceptibility, material exposure and environmental controls.
The most effective sampling plans represent “real life” in the plant, with sampling occurring during hours of normal operation and in locations where materials are exposed to the environment, especially in air drafts. Note that sampling and routine monitoring is only relevant after a system has been validated. For example, an environmental monitoring program should only be established after the HVAC system has been validated.
Documentation: A microbiology expert should examine the facility specifically for microbiological risks. It may be beneficial to conduct a thorough review of a facility’s design, procedures, validation and training annually. An overall risk assessment can be used to document observations, risk profiles and Corrective and Preventative Actions (CaPA). While design improvements are often warranted, procedural mitigation strategies can very often be put in place to help minimize risk. It is important that the technical rationale for these decisions is well documented.
Cleaning: Cleaning is required prior to sanitization. To aid in cleaning, a system should have a smooth surface finish, lack of ridges, minimal breaks/connections, lack of dead-legs and appropriate equipment design. For example, many engineers may choose to use ball valves because they can withstand high pressure; however, most ball valves are not cleanable, as material trapped behind the ball cannot be flushed out of the valve. Butterfly valves are easier to clean and therefore preferable.
Likewise, a system designer may want to use plastic piping such as PVC, however, this is not a cleanable material. Microscopic abrasions in plastic, which may not affect the visible appearance of the surface, can trap material. Even certain grades of stainless steel can be difficult to clean. Whatever design and cleaning procedure choices are made, it is essential to validate cleaning as a separate event from sanitization as described in the previous article.
Sanitization: Sanitization and common sanitizers are discussed in Part 1 of this series. Sanitization validation is essential and typically determined via microbial counts from swabs and/or rinse water samples. It is helpful to consider “worst case” areas of the system when determining sampling locations. This is determined by the experts during a process walkthrough. For example, when employing moist heat sanitization, swab samples should be taken at “cold spots” in the system. Hardest to clean areas such as sharp bends, bolted/flanged connections and tank lid penetration recesses, are informative sample points. Typical success criteria for validation are three consecutive, successful, sanitization runs (as determined by the chosen microbial endpoint). If there is a failure during the validation, the proposed sanitization procedure will need to be amended. Once a sanitization procedure has been successfully validated, a sanitization schedule can be developed that is data-driven and defensible based on risks to the consumer, the product and the manufacturing process. At a minimum, sanitization should occur after each cleaning event.
Facility and Equipment Design: Ideally, facilities are designed with ease of cleaning in mind. Efforts should be made to minimize the complexity of cleaning by trying to make systems cleanable, sanitizable and fully drainable in place. This will affect equipment design choices. Examples of some of the types of considerations that need to be made when designing a facility can be found in the table in this article.
It is recommended that facilities have a logical flow of personnel and materials moving from “dirty” to “clean” areas with appropriate air breaks. Control of environmental airflow (via physical separation and differential pressurization) will help minimize the risk of cross-contamination between “clean” and “dirty” processes. For example, secondary packing often involves manipulation of cardboard and other corrugated materials that generate airborne particles. Laying out a facility such that the free flow of these particles into aseptic areas (e.g. where microbiologically susceptible materials are exposed to the environment) is minimized will reduce the overall microbiological risk for contamination.
Finished Product Quality and Robustness
Monitoring: The CSPA Cleaning Product Division Microbiology Subcommittee has established a Finished Product (FP) method testing guideline to determine if a production run meets microbiological quality specifications as per a general guidance document authored by the same subcommittee.4,5 Limited amount of FP produced can be tested, otherwise there would be no product left to ship to market. Therefore, the holistic, end-to-end control sampling and control plan described in this article contributes to the overall confidence that the testing performed reflects the integrity of every produced unit.
FP testing alone does not compensate for the lack of appropriate controls upstream in the system. Microbial contamination can occur upstream of FP packing, but may not be detected in FP testing. However, given optimal conditions and time, this microbial contamination could spread throughout the system and be detected in FP. Thus, adequate upstream controls combined with suitable FP testing provides the highest level confidence that the system is operating under microbial control.
Documentation: FP testing requirements should be noted on technical standards accessible to all relevant manufacturing plants. This will ensure harmonization across labor shifts, manufacturing sites and regions across the globe. Additionally, documented action plans should exist in case a contamination event is detected, including what action steps should be performed at the plant, who is accountable, and what the ongoing risk is to operations. This documentation can provide consistency as individuals transition from one role to another, and can enable a learning culture that can quickly address similar future issues, should they arise.
Cleaning: For FPs, cleaning can most easily be envisioned as keeping packaging materials clean. It can be very challenging to store and transport packaging with reduced microbiological risks. Enclosed receiving areas at docks, transport lines within the plant, and air blow outs are examples of risk mitigation options. Secondary packaging material, such as cardboard and pallets, present additional microbiological risks. Keeping primary packaging, such as bottles, in sealed transport vessels can help reduce risk during transport, as can the physical and/or procedural separation of primary and secondary packaging processes. It is important to note that packaging materials that require assembly or manipulation present higher levels of risk to be contaminated in the manufacturing environment.
Sanitization: Sanitization frequency of filling equipment can be determined by the overall control state of the system based on overall monitoring results coupled with the FP robustness against microbial contamination. FP robustness is part of a holistic strategy for microbial risk reduction and benefits manufacturing systems by lowering the likelihood of microbial contaminations. A microbial risk assessment of each FP can be performed to determine robustness against potential microbial contamination. More frequent sanitization may be required to reduce risks associated with less robust FP.
Facility and Equipment Design: Filling heads and overflow areas may require wash downs and/or wipe downs to mitigate microbiological risk. Any items used, whether liquid or fabric-based, should be stored appropriately, exposed to the system for the appropriate amount of time, and/or dried completely after use to avoid contamination. Cleaning materials should be stored away from the filling area. Ideally, a filling system can be designed to reduce the need for wipe downs/wash downs.
Architecture of the filling area can also influence microbiological risk. Positive air pressure in the packing area can help reduce risk of incoming bioburden from other areas of the plant. Personnel can be required to take extra preparation steps prior to entering these areas. Also, the items personnel bring into the area, and the time they spend in the area, can be kept to a minimum. A cleaning and sanitization standard can be drafted that includes these specific pieces of information.
Conclusion
CH&I products in general are susceptible to microbial contamination. Therefore effective and holistic microbial control programs are essential in order to deliver safe and efficacious products to market. This article along with Part 1 present the benefits of a systematic approach using five key components of microbial control in manufacturing:
Microbial control is of increasing importance because of trends toward more sustainable products (increased water content and more naturally based raw materials, etc.), which may be more susceptible to microbial contamination. The strategies outlined in this article can be employed to establish a proactive program of controls in the manufacturing process. These controls reduce the potential for microbial contamination of raw materials, systems/facilities and finished CH&I products. Additionally, they are the foundation for effective preservation strategies that maintain microbial integrity during consumer use. Thus, microbial control in manufacturing is an essential component of delivering safe, efficacious and more sustainable products to consumers.
References:
CSPA Microbiology Preservative Subcommittee
The CSPA Microbiology Preservative Subcommittee (MPS) is committed to microbiological quality. To support these goals, a Microbial Control Stewardship Task Force communicates effective preservation strategies. Website: www.cspa.org
Beth Ann Browne • The Dow Chemical Company; Tony Rook • The Sherwin-Williams Company;
Cecilia McGough • Lanxess Corporation
For the Consumer Specialty Products Association Microbiology Preservative Subcommittee
This is the second article of a two-part series dedicated to helping consumer, household and industrial (CH&I) product manufacturers produce products that are uncompromised by microbial contamination (see part 1). CH&I products are often classified as “micro susceptible,” meaning that they are prone to microbial contamination due to a variety of factors that can include compositional features such as high water content and naturally-derived raw materials (RM). In part I of the series, five key components were described for consideration during the CH&I product manufacturing process, including facility and equipment design, cleaning, sanitization, monitoring and documentation.
In this article, these components are further described within the context of the manufacturing process in key areas:
- Supply chain management;
- Raw material management;
- Process design and control; and
- Finished product quality and robustness.
These areas represent the end-to-end model in the manufacturing process. General recommendations along with some specific guidance will be provided for consideration by the reader to improve microbial control in manufacturing systems. CH&I products and the processes to manufacture them are varied and distinct; therefore, the suggestions and tips presented herein may not be directly applicable to all systems and products. It is recommended that the appropriate subject matter experts be consulted in areas such as process engineering, regulatory and microbiology before implementing any manufacturing changes at your facility.
Supply Chain and Raw Material Management
Monitoring: Maintaining microbial integrity of each of the raw materials (RM) is key for effective microbial control of the finished product. A microbiological risk assessment of all RM should be performed to determine material susceptibility to their potential and actual level of microbial contamination. Material sampling, testing and specifications can be established to appropriately manage the risk of a RM lifecycle. Proper RM management programs should be considered as important as finished product testing. When combined, RM, environmental and finished product testing data can establish confidence for product release. Additional information on RM testing can be found in Microbiological Quality of Raw Material General Guideline (MGG 003.1 2014).1
RM control strategies begin with adequate supply chain management of incoming RM. While RM suppliers do not have direct oversight for CH&I product facilities and operations, their attention to RM microbial control will impact the CH&I producing company. A variety of RM suppliers service the CH&I product industry, but they differ notably from one another in their understanding of microbial risks to their materials and contamination potential. For this reason, final product manufacturers must gather sufficient information to effectively assess suppliers. It is common for the CH&I company to conduct a safety and quality inspection of the RM facility prior to final purchase. Supplier monitoring may be accomplished via periodic reviews of documentation and/or physical audits of facilities. During an audit, any issues outlined here can be evaluated and corrective actions can be put into place prior to receipt of the RM. Assessing supply chain logistics is often crucial as it can significantly impact the microbial contamination risk of RM. Examples of these factors include environmental conditions like weather, material transit time (including potential long term storage in transit), transfer equipment and the use of non-dedicated vessels. Certain RM are often considered critical for production or present greater microbial contamination risk; therefore, key suppliers and risk mitigation practices are typically established to protect the supply chain.
Documentation: Typically, materials which are susceptible to microbial contamination are tested by suppliers, and results are added to its certificates of analysis (CoA). A CoA may offer a level of assurance to use the RM without additional microbiological testing, but further risks may be accrued during shipment. Understanding the frequency of microbial testing reported on a CoA by suppliers is also important. A best practice of microbiological quality assurance for the supply chain is to evaluate materials upon receipt and prior to use by finished product producers with proper documentation of test data for traceability, tracking and trending the data.
For any RM testing, proper procedures which include the training of those taking samples should be set. For example, procedures for handling, opening, closing/sealing containers through the sampling process should be in place. Also imperative in the documentation process is the ability to react when a RM is determined to be out of specification. Standard Operating Procedures should be in place to guide these processes.
Cleaning: RM can be delivered in a variety of packaging types. RM that are bulk delivered in reusable containers present different challenges than those supplied in disposable packaging. Even drums are frequently resused. Single-use containers should be used whenever feasible and practical. Proper procedures for maintaining reused packing are critical for microbial control from the RM supplier location to the manufacturing facility. Reused containers dedicated to a specific RM are preferred; however, RM can be frequently transported in bulk quantities in reusable containers shared with other materials. Proper cleaning of containers, according to a validated procedure, is needed between uses to maintain the quality of the material, and documentation of the process should be available upon receipt of material(s). Within manufacturing systems, cleaning of RM storage handling equipment is vital. Many RM, such as silicones, can build up within the manufacturing system. RM systems that are not properly cleaned create environments conducive to microbial growth, raising risks for microbial contamination.
Sanitization: RM may be transported and stored in a variety of conditions, and decisions must be made about the frequency of sanitization of the storage vessels, and the maximum amount of time allowed between sanitization and vessel filling. Large reusable transport vessels should be sanitized prior to being filled with a microbiologically susceptible RM. However, not all vessel types can be sanitized, such as lined rail cars, which require other risk mitigation methods to be identified and implemented, such as confined entry inspection to verify cleaning.
Documentation of the sanitization process or alternative risk mitigation steps should be included in the delivery documentation. Upon receipt at the manufacturing site, some materials may be stored for extended periods in the shipped vessels while other RM may be transferred into large storage tanks. Sanitization frequencies can vary based on how long the RM is exposed to the system. For example, a RM with rapid turnover in storage systems would pose different risks than the same material stored for long periods with infrequent use.
Facility and Equipment Design: Upon receipt, RM must be offloaded at the manufacturing site. Specific procedures should be in place for this process, noting that some materials will need special considerations. It is preferred to use in-house offloading hoses instead of transporter hoses for liquids as the recipient has greater control over proper cleaning, sanitization and storage of the in-house hoses between uses. Likewise, the location for unloading bulk samples needs adequate protection for microbial control.
RM quality within the manufacturing site after offloading encompasses a large portion of the manufacturing process, and the fundamentals of process control and design outlined in the next section will cover precautions for equipment used in conjunction with RM. It should be noted that exterior storage of RM, often done because of large usage volumes, is an area that requires special considerations, but that area is beyond the scope of this article.
Process Design & Control
Monitoring: It is essential to monitor manufacturing systems to ensure they are in control. Typical programs may include monitoring of environmental air, bulk storage tanks, in-process mixes, etc. Regulatory bodies give little to no prescriptive guidance on breadth or frequency of sampling and monitoring. Rather, it is left to the company to develop a sampling plan that is sufficient to demonstrate consistent control of each system. However, USP <1116> Microbiological Control and Monitoring of Aseptic Processing Environments,2 and USP <1231> Water for Pharmaceutical Purposes3 are good resources to learn about principles that should be considered when developing monitoring programs. The same types of considerations should be taken into account for all monitoring programs including those for bulk storage tanks and in-process equipment. Some factors may include microbiological susceptibility, material exposure and environmental controls.
The most effective sampling plans represent “real life” in the plant, with sampling occurring during hours of normal operation and in locations where materials are exposed to the environment, especially in air drafts. Note that sampling and routine monitoring is only relevant after a system has been validated. For example, an environmental monitoring program should only be established after the HVAC system has been validated.
Documentation: A microbiology expert should examine the facility specifically for microbiological risks. It may be beneficial to conduct a thorough review of a facility’s design, procedures, validation and training annually. An overall risk assessment can be used to document observations, risk profiles and Corrective and Preventative Actions (CaPA). While design improvements are often warranted, procedural mitigation strategies can very often be put in place to help minimize risk. It is important that the technical rationale for these decisions is well documented.
Cleaning: Cleaning is required prior to sanitization. To aid in cleaning, a system should have a smooth surface finish, lack of ridges, minimal breaks/connections, lack of dead-legs and appropriate equipment design. For example, many engineers may choose to use ball valves because they can withstand high pressure; however, most ball valves are not cleanable, as material trapped behind the ball cannot be flushed out of the valve. Butterfly valves are easier to clean and therefore preferable.
Likewise, a system designer may want to use plastic piping such as PVC, however, this is not a cleanable material. Microscopic abrasions in plastic, which may not affect the visible appearance of the surface, can trap material. Even certain grades of stainless steel can be difficult to clean. Whatever design and cleaning procedure choices are made, it is essential to validate cleaning as a separate event from sanitization as described in the previous article.
Sanitization: Sanitization and common sanitizers are discussed in Part 1 of this series. Sanitization validation is essential and typically determined via microbial counts from swabs and/or rinse water samples. It is helpful to consider “worst case” areas of the system when determining sampling locations. This is determined by the experts during a process walkthrough. For example, when employing moist heat sanitization, swab samples should be taken at “cold spots” in the system. Hardest to clean areas such as sharp bends, bolted/flanged connections and tank lid penetration recesses, are informative sample points. Typical success criteria for validation are three consecutive, successful, sanitization runs (as determined by the chosen microbial endpoint). If there is a failure during the validation, the proposed sanitization procedure will need to be amended. Once a sanitization procedure has been successfully validated, a sanitization schedule can be developed that is data-driven and defensible based on risks to the consumer, the product and the manufacturing process. At a minimum, sanitization should occur after each cleaning event.
Facility and Equipment Design: Ideally, facilities are designed with ease of cleaning in mind. Efforts should be made to minimize the complexity of cleaning by trying to make systems cleanable, sanitizable and fully drainable in place. This will affect equipment design choices. Examples of some of the types of considerations that need to be made when designing a facility can be found in the table in this article.
It is recommended that facilities have a logical flow of personnel and materials moving from “dirty” to “clean” areas with appropriate air breaks. Control of environmental airflow (via physical separation and differential pressurization) will help minimize the risk of cross-contamination between “clean” and “dirty” processes. For example, secondary packing often involves manipulation of cardboard and other corrugated materials that generate airborne particles. Laying out a facility such that the free flow of these particles into aseptic areas (e.g. where microbiologically susceptible materials are exposed to the environment) is minimized will reduce the overall microbiological risk for contamination.
Finished Product Quality and Robustness
Monitoring: The CSPA Cleaning Product Division Microbiology Subcommittee has established a Finished Product (FP) method testing guideline to determine if a production run meets microbiological quality specifications as per a general guidance document authored by the same subcommittee.4,5 Limited amount of FP produced can be tested, otherwise there would be no product left to ship to market. Therefore, the holistic, end-to-end control sampling and control plan described in this article contributes to the overall confidence that the testing performed reflects the integrity of every produced unit.
FP testing alone does not compensate for the lack of appropriate controls upstream in the system. Microbial contamination can occur upstream of FP packing, but may not be detected in FP testing. However, given optimal conditions and time, this microbial contamination could spread throughout the system and be detected in FP. Thus, adequate upstream controls combined with suitable FP testing provides the highest level confidence that the system is operating under microbial control.
Documentation: FP testing requirements should be noted on technical standards accessible to all relevant manufacturing plants. This will ensure harmonization across labor shifts, manufacturing sites and regions across the globe. Additionally, documented action plans should exist in case a contamination event is detected, including what action steps should be performed at the plant, who is accountable, and what the ongoing risk is to operations. This documentation can provide consistency as individuals transition from one role to another, and can enable a learning culture that can quickly address similar future issues, should they arise.
Cleaning: For FPs, cleaning can most easily be envisioned as keeping packaging materials clean. It can be very challenging to store and transport packaging with reduced microbiological risks. Enclosed receiving areas at docks, transport lines within the plant, and air blow outs are examples of risk mitigation options. Secondary packaging material, such as cardboard and pallets, present additional microbiological risks. Keeping primary packaging, such as bottles, in sealed transport vessels can help reduce risk during transport, as can the physical and/or procedural separation of primary and secondary packaging processes. It is important to note that packaging materials that require assembly or manipulation present higher levels of risk to be contaminated in the manufacturing environment.
Sanitization: Sanitization frequency of filling equipment can be determined by the overall control state of the system based on overall monitoring results coupled with the FP robustness against microbial contamination. FP robustness is part of a holistic strategy for microbial risk reduction and benefits manufacturing systems by lowering the likelihood of microbial contaminations. A microbial risk assessment of each FP can be performed to determine robustness against potential microbial contamination. More frequent sanitization may be required to reduce risks associated with less robust FP.
Facility and Equipment Design: Filling heads and overflow areas may require wash downs and/or wipe downs to mitigate microbiological risk. Any items used, whether liquid or fabric-based, should be stored appropriately, exposed to the system for the appropriate amount of time, and/or dried completely after use to avoid contamination. Cleaning materials should be stored away from the filling area. Ideally, a filling system can be designed to reduce the need for wipe downs/wash downs.
Architecture of the filling area can also influence microbiological risk. Positive air pressure in the packing area can help reduce risk of incoming bioburden from other areas of the plant. Personnel can be required to take extra preparation steps prior to entering these areas. Also, the items personnel bring into the area, and the time they spend in the area, can be kept to a minimum. A cleaning and sanitization standard can be drafted that includes these specific pieces of information.
Conclusion
CH&I products in general are susceptible to microbial contamination. Therefore effective and holistic microbial control programs are essential in order to deliver safe and efficacious products to market. This article along with Part 1 present the benefits of a systematic approach using five key components of microbial control in manufacturing:
- Facility and equipment design
- Cleaning
- Sanitization
- Monitoring
- Documentation
Microbial control is of increasing importance because of trends toward more sustainable products (increased water content and more naturally based raw materials, etc.), which may be more susceptible to microbial contamination. The strategies outlined in this article can be employed to establish a proactive program of controls in the manufacturing process. These controls reduce the potential for microbial contamination of raw materials, systems/facilities and finished CH&I products. Additionally, they are the foundation for effective preservation strategies that maintain microbial integrity during consumer use. Thus, microbial control in manufacturing is an essential component of delivering safe, efficacious and more sustainable products to consumers.
References:
- Micrbiological Quality of Raw Material General Guidelines MGG 003.1. CSPA Cleaning Products Divison Test Methods and Guidelines Compendium; Fifth Edition; 2012.
- 2016 U.S. Pharmacopoeia-National Formulary [USP 36]. Volume 1. Rockville, Md: United States Pharmacopeial Convention, Inc; 2015. <1116> Microbiological Control and Monitoring of Aseptic Processing Environments
- 2016 U.S. Pharmacopoeia-National Formulary [USP 36]. Volume 1. Rockville, Md: United States Pharmacopeial Convention, Inc; 2015. USP <1231> Water for Pharmaceutical Purposes
- Microbiological Examination of Household and Institutional Products: Method Guideline for Microbial Enumeration Tests MMG 001.1.CSPA Cleaning Products Divison Test Methods and Guidelines Compendium; Fifth Edition; 2012.
- Microbiological Quality of Household and Institutional Products General Guidelines MGG 002.1. CSPA Cleaning Products Divison Test Methods and Guidelines Compendium; Fifth Edition; 2012.
CSPA Microbiology Preservative Subcommittee
The CSPA Microbiology Preservative Subcommittee (MPS) is committed to microbiological quality. To support these goals, a Microbial Control Stewardship Task Force communicates effective preservation strategies. Website: www.cspa.org